Introduction
scRNA-seq 数据的计算分析包括质控,比对,定量,标准化,聚类,轨迹分析以及差异表达基因的鉴定等,其中聚类是给予转录组数据定义细胞类型的一个核心步骤,也是 scRNA-seq 最广泛的应用之一。基于转录组数据来确定细胞类型是一种数据驱动的、无偏差的方法,推动了多个大型图谱项目的出现,致力于构建不同物种,不同器官,不同发育阶段所有细胞的完整参考集,从而加深我们对生物学的认知。虽然近年来涌现出大量针对单细胞数据的聚类算法,但是对到底哪种更好或者如何基于 scRNA-seq 数据定义细胞类型仍未达成共识。本综述中,作者将从现有的聚类方法,无监督聚类相关的挑战,对结果的生物学解释以及未来聚类方法的进化方向等方面入手展开讨论
现有的聚类算法
许多聚类算法具有普适性,可用于任何类型的数据,因为他们以计算各个样本间的距离作为聚类的基础。但是对于 scRNA-seq 数据,由于检测了大量的基因,因此在得到的高维空间内,数据点(或者细胞)之间的距离是很相近的,这一现象被称为“维度灾难”(curse of dimensionality)。由于该现象的存在,我们没办法在高维空间通过计算距离将细胞分群,所以出现两类方法用于降低噪音,加速计算:特征选择(feature selection)和降维(dimendionality reduction),其中特征选择是指找出包含信息最多的基因,例如那些方差最大的,而降维则是将数据映射至低维空间,比如 PCA。之后,在低维空间或者仅基于筛选出的基因计算距离。同样,距离的计算方式也有多种选择,包括 Euclidean distance,cosine similarity,Pearson’s correlation 和 Spearman’s correlation 等。其中后三种方法的主要优势是他们的维度不变性,即考虑的是数据之间的相对差异,因此对 library 或者 cell size 差异更加 robust
最受欢迎的聚类算法是 k-means,它通过不断迭代找出 k 个中心,然后将各个细胞分配至最近的中心点。标准 k 均值方法,称为 Lloyd’s algorithm,它的计算复杂度为线性,因此可用于大型数据集;但是 k-means 是一个贪婪算法,无法确保找到整体上的最优结果。这一不足可通过像 SC3 那样使用不同的初始值多次进行 k-means 计算,最终找到一致结果来克服。k_means 的另一个缺点是偏向于得到大小相同的 cluster,这可能导致罕见细胞和其他的细胞群合在一起。为解决这一问题,RaceID 增加了异常值检测来鉴定罕见细胞类型,而 SIMLR 则是在进行 k-means 计算的同时训练一个自定义的距离衡量值
另一个常用的聚类算法是层次聚类,它能够循序地不断将单个细胞整合成大的分支(agglomerative)或将分支分成更小的组(divisive)。该算法的一个主要缺点是其时间和内存消耗均至少为平方增长型,因此难以用于大型数据集。CIDR 在计算距离时增加了对零值的隐式推断,这样在低深度样本中可以得到更稳定的细胞-细胞距离计算值;还有一些算法如 BackSPIN 和 pcaReduce,在每次融合或切分之后都进行降维,通过这样的迭代方法来提升对小分支的鉴定能力
由于前两种方法的局限性,基于社区发现(community-detection-based)的算法得到了越来越多的应用。社区发现是聚类的一种变型,专门用于图。不同于其他方法是找出相近的点,社区发现是找出密集相连的点。要将该方法用于 scRNA-seq 数据,首先需构建 k 最近邻居图,其中 k 的选择会影响最终分支的数量和大小。为了加强对异常值的鲁棒性,通常还会基于每对细胞的共享最近邻居进行重加权。与层次聚类返回所有 level 上的分区不同,绝大部分基于图的方法只返回单个结果,从而使得运行更快。绝大部分基于图的方法不需要用户指定分支的数目,而是采用间接性的分辨率参数,比如增加邻居数,将导致分辨率降低,即分支减少。Louvain community detection algorithm 是目前这些算范中应用于 scRNA-seq 数据最广泛的一种,其中 PhenoGraph 最早使用了 shared-nearest-neighbour graphs 和 Louvain community detection,后来这一联合方法也被整合进了 Seurat 和 scanpy
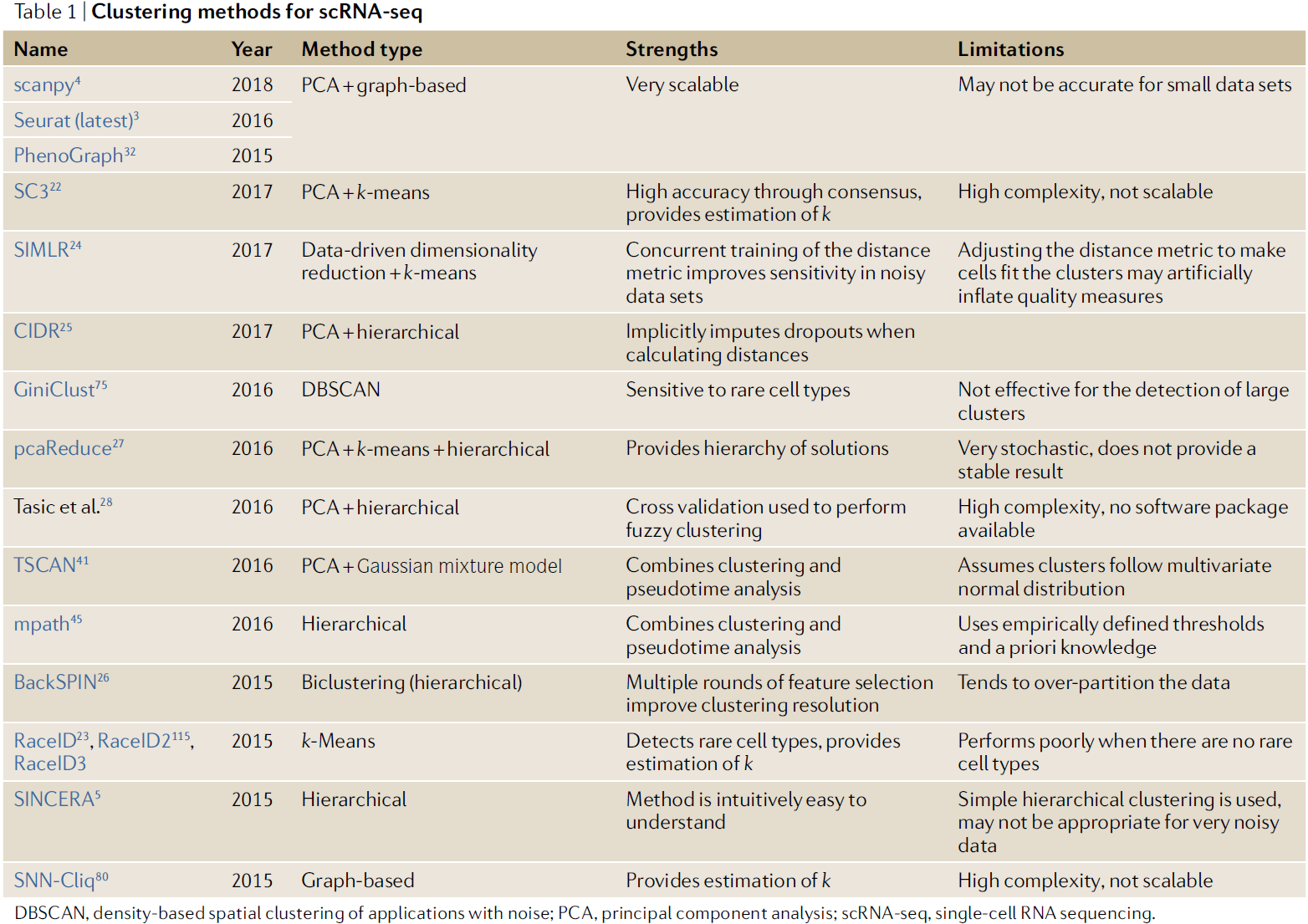
目前已经出现了不少用于 scRNA-seq 数据的聚类方法(下图),其中 scanpy 和 Seurat 包中的聚类方法受众最广,但也有研究表明基于 Louvain 算法的聚类方法在小数据集上的效果不佳。不同的方法有其适用性,需要根据实际情况进行选择
离散还是连续的细胞分组
现有大部分算法会将数据进行划分,但不考虑这些分组是否具有生物学意义,往往还会将随机噪音误认为是单独的分组。所以,当实际并不存在离散的分组时,聚类可能不是一个正确的选择,例如考虑发育轨迹时,我们需要将细胞沿着一条连续的轨迹放置,即拟时序(pseudotime)。有些作者开发了新的策略来串联起拟时序和聚类方法,如 Tasic et al 通过 bootstraping 策略将细胞划分为稳定分配为相同分组和分配为不同分组的两类,标记为稳定的和瞬时的,其中后者可能是处于中间态的细胞群;还有像 Wolf et al,采用 coarse-grained graph representation,将细胞分配到节点(表明稳定组)或边上。在实际分析中,我们可能预先不清楚到底是分群还是轨迹,可以考虑两类方法都尝试,可能会带给我们不同角度的信息
技术性挑战
由于初始 RNA 含量较少,scRNA-seq 数据呈现出更多的噪音以及更多的零值,因而给分析也带来了更多的挑战。目前有一些统计方法可以对零值进行推算,但是需要基于已知的细胞-细胞或基因-基因关联
由于每个细胞是生物学,而不是技术重复,因此给预估单细胞数据中的技术噪音也带来了挑战,目前也开发了一些基于外源 RNA 的噪音模型,可以通过添加模拟的噪音然后进行重聚类来评估分支的鲁棒性
批次效应(batch effect)是一类由于实验因素引起的基因表达量改变,比如不同的操作人或者测序仪中不同的泳道,会对聚类结果产生较大的影响。避免批次效应的最好方法是采用平衡的实验设计,使得样本划分到不同的试验批次,从而估算并剔除批次效应,但有些情况下可能无法达到
此外如何处理样本也会带来巨大的影响,比如尸体样本,RNA 可能发生可不均匀的降解;还有像在分离敏感的组织,如神经元细胞时,可能会活化早期基因或其他应激基因的表达。目前采用的策略包括添加抑制剂,冰冻保存或者化学固定等,但总的来看还得对 protocol 继续优化
有时候像 doublets,可能呈现出中间态的表现,却被误认为是单独的分支,现在有一些基于平板或者微流芯片的 protocol,可以在裂解前对捕获的细胞成像,鉴别是否存在 doublets,此外还有一些算法可以进行推断
总的来讲,由于存在大量的干扰因素,对于像聚类结果,需要去检验线粒体 RNA,试验批次,测序深度以及检测到的基因数目等是否是导致分支的原因,从而确保每个分支是具有生物学意义的
生物学挑战
除了技术噪音,瞬时的生物学状态可能也会掩盖真实的细胞身份,比如细胞周期,目前 scLVM 和 cyclone 等能够用于剔除细胞周期效应,校正转录组。但在有些情况下,这可能确实是与细胞类型相关,因此不应剔除,类似的还有如总 RNA 含量等。所以,还是需要根据实际的情况来进行判定
组织内不同类型细胞占比的巨大差异也给聚类带来了挑战,不同类型细胞占比可能相差两个数量级,而许多聚类算法是在各分支大小相近的情况下表现最好。 GiniClust 和 RaceID 等专为鉴别罕见细胞类型而定制,遗憾的是这影响了它们在鉴定常见细胞类型时的表现。有一些作者采取了分而治之的策略,先初步聚类得到一个大一点的分支,然后再对分支进一步聚类,不过如何确定一个分支是否需要重聚类也是个挑战
计算挑战
许多 scRNA-seq 数据集很大,一方面这可以增强统计效力,找到罕见的细胞类型,另一方面也给计算分析带来了巨大的挑战。由于大量的 droupout 和噪音,线性转换方法如 PCA 无法准确捕捉细胞间的关系,而非线性方法会更加灵活,得到的结果往往更好看。常用的非线性降维方法包括 tSNE 和 UMAP,不过这些方法主要的限制是包含需要用户手动确定的参数,而这些值对可视化结果影响很大。有些是直接敲定分支数,有些则是确定最近邻居数来间接影响聚类结果,目前也存在一些方法来指导参数的选择,比如基于聚类质量分数,或基于拐点,具体还是得根据数据情况,比如想寻找罕见细胞,那么需要增加分支数
除此之外,如何验证计算分析结果可能是整个分析中最具挑战性的一步。验证方法的可靠性可以使用模拟的数据集,但是这些数据可能无法完全体现真实数据的复杂性。其他的策略可以使使用像 seqFISH,RNAscope 和 merFISH 等方法来进行验证,但这类方法的分辨率有限
生物学注释
聚类之后需要我们确认每个分支的生物学意义,这是一个费时的过程。由于没有固定的规则指导如何定义一个细胞类型,也没有包含所有细胞及特征的数据库,所以很多时候取决于研究人员自身掌握的背景知识。一般情况下大家得到的结果其实会比较接近,但这也可能是因为受到现有文献的偏差影响
随着数据越来越多,一种新的策略是将新鉴定的分支与已注释好的数据进行比较,但是批次效应的存在给这一过程带来了挑战,目前主要的两种策略是混合(merging)和映射(projection)。如果两个样本具有相似的生物学来源,可以考虑在聚类之前将数据融合,不过需要消除批次效应的影响。另一种方法是将一个数据集中的细胞映射到另一个数据集上,基于与 query 细胞最相近的细胞的注释来进行聚类等,适用于其中一个数据集很大的情况,不过假如存在新的细胞类型,可能导致聚类和注释失败,所以关键是要有一个全面的数据图谱库
还有一个策略是构建 cell ontology 数据库,基于基因表达,以合理且全面的方式描述细胞状态以及相互之间的关系。与 gene ontologies 相似,cell ontologies 也是分层的,且能确保注释的一致性
总结
无监督聚类作为 scRNA-seq 分析中的核心步骤,是许多下游分析的基础,因此对最终结果产生重大影响。不同的算法可能有其最适用的场景,未来也可能出现新的算法,具体的选择还是需要基于实际的数据进行判断。未来随着数据量的不断增加,多组学数据的整合,以及空间信息的加入,对聚类也会带来新的挑战。除了计算分析层面,生物学注释也很重要,需要社区间的合作以确保更好的一致性
REF
- Kiselev VY, et al. Challenges in unsupervised clustering of single-cell RNA-seq data. Nat Rev Genet, 2019, 20(5):273-282. doi: 10.1038/s41576-018-0088-9.