Introduction
scRNA-seq 数据分析相关计算方法的快速发展为我们的工作带来了更多的选择,但同时也需要我们对这些方法进行系统的比较以明确不同算法的优劣势。虽然已经有一些研究针对各个分析步骤,如标准化、特征选择、差异表达分析、聚类以及轨迹分析等相关的方法进行比较,但是存在两个问题:一是他们选择的模拟数据集预设的假设可能无法匹配实际 scRNA-seq 数据;二是他们针对特定步骤,而不是在整个分析流程这一层面。因此,作者设计了一系列实验,混合来自不同细胞系的细胞或 RNA,并通过梯度稀释模拟不同细胞内 RNA 含量的差异,另外还加入 ERCCs 等,之后通过不同 scRNA-seq 平台获取数据,并基于这些数据集对整个分析流程各步骤相关软件进行比较
实验设计
实验设计部分包含:
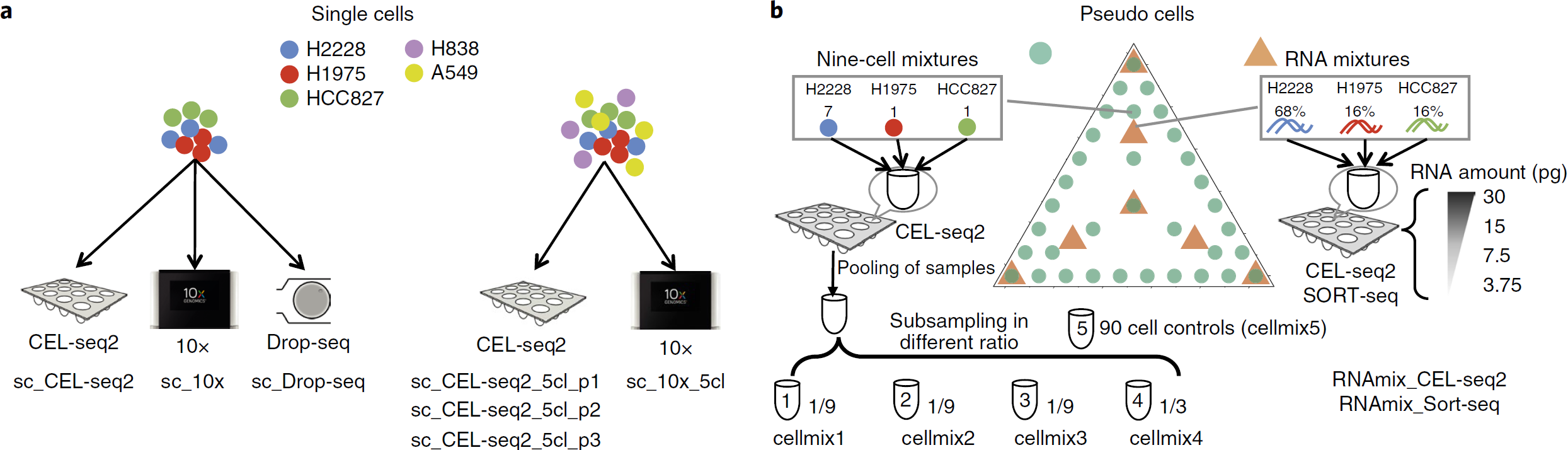
- single cell 层面(下图a):总共5个人类肺腺癌细胞系(HCC827, H1975, A549, H838 and H2228),分为两种情况:
- 以相同的比例混合其中3个细胞系(HCC827, H1975 and H2228)并用 CEL-seq2、10x 和 Drop-seq 分别测1组
- 以相同比例混合所有5个细胞系并用 CEL-seq2 测3个重复,10x 测1份
共计7组。对于每个细胞系,其身份可以根据已知的遗传变异推断。该设计主要可比较不同 protocol 的影响,以及数据整合方法的效果
- RNA mixture 层面(下图b中黄色三角形):从 HCC827, H1975 and H2228 细胞群中提取 RNA,以不同比例混合并稀释到单细胞水平的量(所以每个孔可视为单个细胞),总计8种不同混合物(mix1-8),其中 mix1 和 mix2 均为三种细胞各占1/3,但是是分开处理的,可用于评估 RNA 稀释以及混合过程中引入的差异。除了不同比例的混合外,作者还设计了一系列稀释梯度,添加的 RNA 数量为3.75,7.5,15和30pg,每组混合物在每块板上各重复6,14,14,14次(所以正好一块384孔板),从而模拟细胞大小差异。之后分别用 CEL-seq2 和 SORT-seq 测序,故得到2个数据集。RNA mixture 数据集中包含技术重复,梯度稀释,因此适合比较标准化和填补方法
- cell mixture 层面(下图b绿色圆圈):将 HCC827, H1975 and H2228 细胞分选至384孔板。设计的 major tracjectory (中间的三叉型)类似于上面的 RNA mixture,每种组合(共10种)是9个细胞,不同细胞系所占比例不同,构成“拟轨迹”,一块板上每种组合各20个重复。对于主轨迹,每种组合还配了一个90个细胞的群体对照,3个重复。minor tracjectory (三角形的边)上,则是两种细胞系的不同比例组合,共24种,5个重复。此外,还加了9组3:3:3组合的低质量细胞重复,9组1:1:1组合模拟小细胞的重复。除了用于轨迹分析,通过改变细胞数和细胞质量研究这些对数据特征的影响。含有9个细胞的孔混合后进行了抽样以得到单细胞的量。其中3个重复取1/9,1个重复取1/3。对3个重复采用了不同比例的 clean-up,从而产生完全由于技术因素导致的批次效应,并可以研究 clean-up 对数据的影响
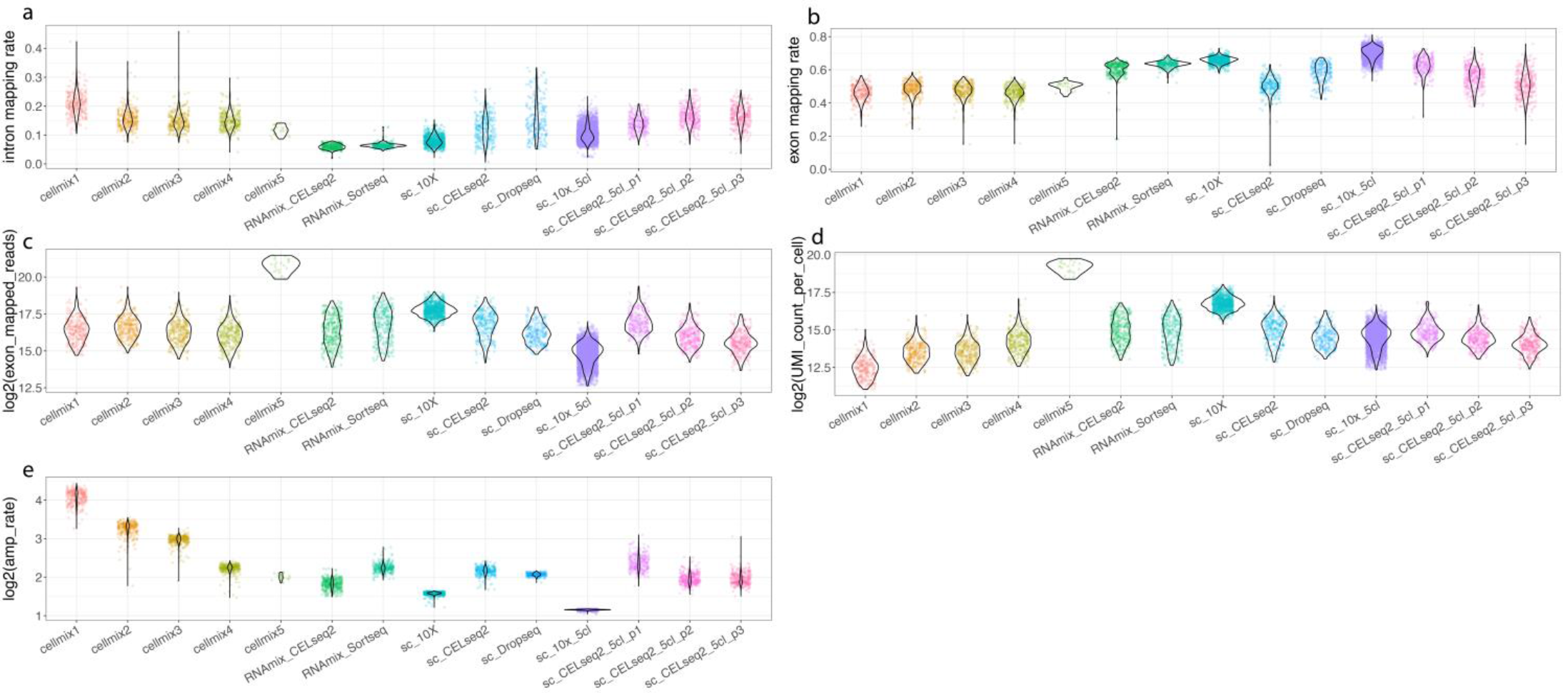
质控指标检验
利用 scPipe 包获得各个数据集的一系列质控指标,发现来自不同平台的数据在外显子比对率和每个细胞总 UMI 数方面表现出一致的高质量,但在内含子比对率上呈现较大差异(下图)。利用 scran 标准化,并绘制 PCA 图,可以看到显示的结构与预期的比较一致。另外不同数据集 doublet 率不同
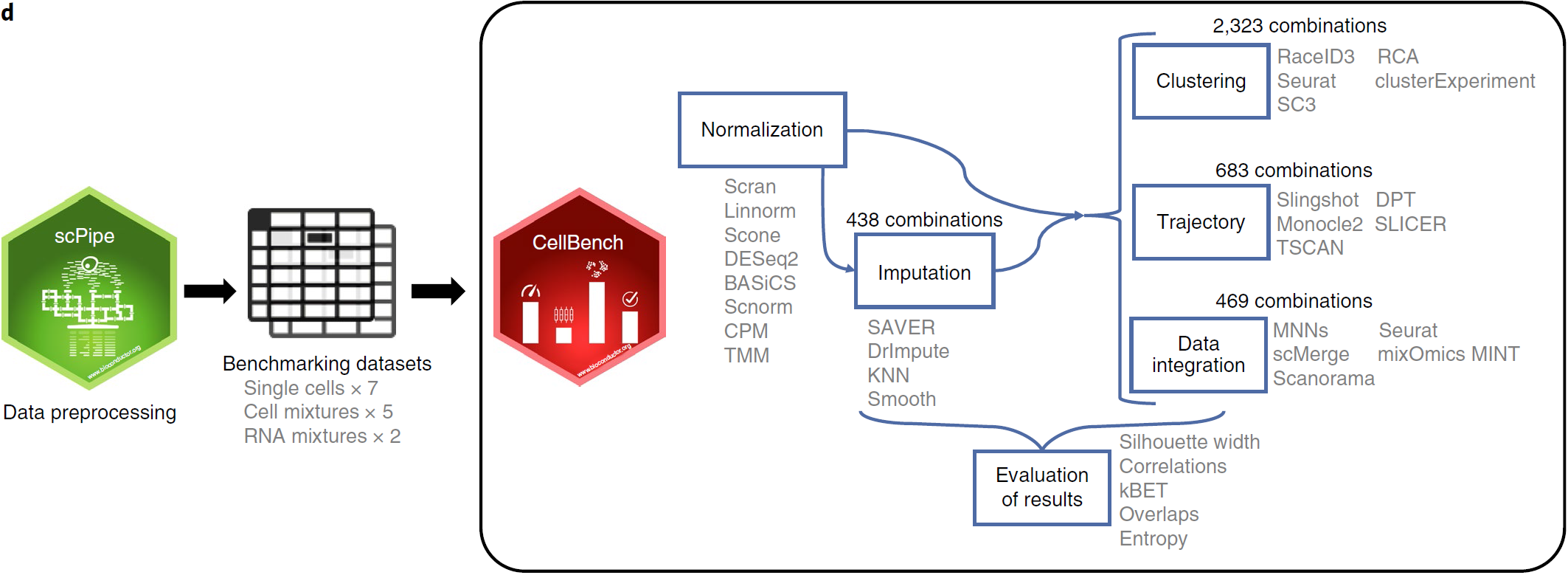
整个 workflow 如下图所示,经过预处理和初步质控,每个数据集被不同的方法组合分析,包括8种标准化方法,3种填补方法,7种聚类方法,5种轨迹分析方法以及6种数据整合方法
标准化和填补方法的比较
选择的标准化方法共8种,包括:
- 主要针对 bulk 开发的:trimmed mean of M-values (TMM), count-per-million and DESeq2
- 针对 scRNA-seq 的:scone, BASiCS, SCnorm, Linnorm and scran
填补方法有3种:kNN-smoothing, DrImpute and SAVER。总计数据集+标准化+推断的组合为438个
采用2种指标评估方法的优劣:
- silhouette width of cluster (for all datasets)
- Pearson correlation coefficient of normalized gene expression within each group (for RNA mixture data)
发现除了 TMM,所有标准化方法均能提高 silhouette width,且针对 scRNA-seq 数据开发的方法表现更好,不过 DESeq2 也给出了比较好的结果。在所有方法中,Linnorm 表现最佳,scran 和 scone 紧随其后
RNA mixture 实验中的“拟细胞”使用不同数量的初始 RNA 来模拟 droupout,使得它成为比较填补方法的理想数据集。通常,填补方法会导致组内相关性提升,但会因标准化方法的不同产生差异。kNN-smoothing 和 DrImpute 与不同标准化方法的组合比较得到的结果比较一致,而 SAVER 变化较大
填补可能导致出现伪造的细胞,为了研究该情况,作者选择了 RNA mixture 数据集中仅含 H2228 或 HCC827 细胞的样本,比较相关性。整体来看,同一组内,样本间相关性随着 mRNA 含量减少而降低(下图g),但通过填补后,相关性会提升(下图h,i和j)。3种填补方法都能够清晰地区分出两组细胞,但是 kNN-smoothing 会引入一个假的组内关联结构(下图h),而且额外的分支与 RNA 含量相关,表明 kNN-smoothing 对 droupout 敏感
聚类方法的比较
作者选取了5种代表性的聚类方法用于所有的数据集,包括:
- RaceID3
- RCA
- Seurat
- clusterExperiment
- SC3
为了评估不同聚类方法的表现,计算以下2个指标,用于评估是否存在过聚类或低聚类:
- entropy of cluster accuracy (ECA)
- entropy of cluster purity (ECP)
其中具有较低的 ECP 和较低的 ECA 值对应最佳的聚类结果。这两项指标的与 adjusted Rand index(ARI)具有较好的一致性,而后者是一种常用的衡量聚类效果的方法。结果显示默认的参数设置下,没有哪一种方法在所有实验设计中都能表现最优。总的来说,Seurat 能够较好地平衡过聚类和低聚类之间的关系,RaceID3 在相对更复杂的细胞混合数据集中表现更佳。不过由于该类数据集存在连续性的群体结构,使得分支间的分离度相较其他数据集更小,导致所有的方法在该数据集上的准确性都要比在其他数据集上差
在单细胞数据集上,基于基因型信息得到的分支数量可能低估了实际值,这些细胞系中可能存在具有微小差异的亚分支,所以那些在该数据集上过聚类的方法可能是捕获到了真实的生物学信号
作者以 ARI 或真实的分支数量为因变量,不同的方法作为协变量,用线性模型拟合2323种分析组合(数据集+标准化+填补+聚类),进一步探索每种方法对结果的贡献,发现 clusterExperiment 方法经常恢复出预期的群体结构,而 SC3 会低聚类。kNN-smoothing 与低 ARI 和高分支数量相关,暗示该方法会引入假分支
轨迹分析方法的比较
作者比较了5种方法用于 RNA mixture 和 cell mixture,包括:
- Slingshot
- Monocle2
- SLICER
- TSCAN
- DPT
mixture 数据集通过改变 cell 或 RNA 比例模拟出“轨迹”。为简单起见,将 H2228 作为轨迹的根,比较各方法得到的拟时序与实际之间的相关性,此外还计算了每条轨迹的覆盖率,即有多少比例的细胞是被分配到正确的轨迹上。总计683种分析组合(标准化+填补+轨迹分析)被用于各个数据集。此外,利用线性模型量化每种方法的平均表现,发现根据2项指标,Slingshot 和 Monocle2 的结果足够 robust,且产生的轨迹有对应的生物学意义,不过 Slingshot 偶尔会给出额外的轨迹。相反,SLICER 能将所有细胞放置在正确的路径上,但是没办法将它们排好序或者恢复出预想的结构。虽然 Slingshot 和 Monocle2 表现差不多,但是它们放置细胞的方式存在差异。Slingshot 自己不进行降维,而 Monocle2 利用 DDR-tree 降维且倾向于将细胞放置在树的节点上而不是节点间,这一特征可能更适应实际数据只能够位于中间态的细胞数量较少的情况,但是这个前提也不总是成立,所以在解释结果的时候需要注意
数据整合方法的比较
作者比较了5种方法用于单细胞和 RNA mixture 数据集,包括:
- MNNs
- Scanorama
- scMerge
- Seurat
- MINT
其中5个细胞系混合的 CEL-seq2 数据集由于 doublet 率过高被排除。469种组合(标准化+填补+整合)应用于两组实验设计。与预期的一样,直接把独立的数据集放在一起时,PCA 图上可以观察到不同 protocol 之间明显的分离,而数据整合方法能够不同程度地消除这些效应。MNNs,Scanorama 和 scMerge 会返回校正批次之后的表达矩阵,可用于其他下游分析工具,而 Seurat 和 MINT 输出的是数据在低维空间的表示。通过计算 protocol(batch)和已知的细胞系或混合物组信息的 silhouette width 来评估各方法的表现。由于 Seurat 使用 t-SNE 降维,导致距离信息未能保留,因此使用 kBET 量化残留的批次效应差异。在单细胞数据集上,大部分方法表现差不多,除了 Seurat 的 kBET acceptance rate 值较低;而在囊括了更多组别,且每组细胞数量更少的 RNA mixture 实验中,各方法的表现差异较大,其中 MNNs 在 silhouette width distance 上最好,而 Seurat 在 kBET acceptance rate 上最高。除了 silhouette width 和 kBET acceptance rate 外,还对整合后的 RNA mixture 数据进行了聚类,发现除了 Seurat,大部分方法在正确标准化和填补之后,具有较高的 ARI,给出了正确地分支数量,表明它们能够保留好生物学信号
Discussion
通过各式各样的组合,作者对数据分析各步骤的不同方法的表现进行比较,发现各方法在不同数据集上的表现存在差异,没有一个适用于任何情形的方法,但是像标准化和填补中的 scran,Linnorm,DrImpute 和 SAVER,聚类中的 Seurat,轨迹分析中的 Monocle2 和 Slingshot,以及数据集整合中的 MNNs 始终能给出令人满意的结果。一些标准化和填补方法的组合,如 Linnorm+SAVER,其大部分下游分析结果都比较好。各方法处理不同输入的能力也存在差异,像 Linnorm 和 SC3,不管输入数据是什么,结果都相对稳定,而 SAVER 则对输入比较敏感
通过比较各方法组合在不同任务中的结果,作者发现一系列关于不同预处理方法对下游分析的适用性的有趣趋势,比如虽然填补能够提升聚类和轨迹分析的表现,但是会导致数据整合分析中不同批次数据混合效果较差
一些综合性方法能够整合多种算法来提升表现,但是并不总是比单一方法来得好,比如 SC3 和 clusterExperiment 并没有超过其他聚类方法,scone 标准化在不同数据集给出的结果也很混杂
REF
- Benchmarking single cell RNA-sequencing analysis pipelines using mixture control experiments. Nat Methods, 2019, 16(6):479-487. doi: 10.1038/s41592-019-0425-8. Epub 2019 May 27.