Smart-seq2 (2013)
Smart-seq2 的流程如下图所示:
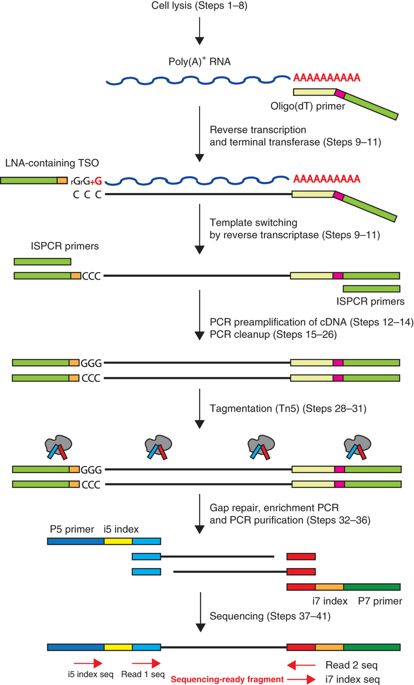
- 细胞裂解。使用相对较温和的方式(低渗溶液)裂解细胞。裂解液中包含了 free dNTPs 和具有 oligo-dT 的寡核苷酸序列(包含30nt的 oligo-dT 和25nt的通用5’锚定序列),后者能够起始具有 polyA 尾的 RNA 的 RT 反应。free dNTPs 能够提高 RT-PCR 的产量。
- RT。当 RT 进行至 RNA 5’端时(合成是从5’-3’,对 RNA 模版而言是从其3’-5’),会在合成的 cDNA 3’ 末端添加2-5个 untemplated nucleotides,而模版转换反应(template-switching reaction)正依赖于这几个碱基。TSO(template-switching oligos)3’末端会携带与这几个碱基互补的序列,以及与 oligo-dT 引物相同的锚定序列(使得后面的 PCR 只需要使用单种引物),与RNA 5’端连接,然后反转录酶实现模版转换,在 cDNA 3’端合成与 TSO 互补的序列。
- 预扩增。当合成第一条链后,进行有限次数的循环,得到的材料足够进行后续分析即可。
- Tagmentation。对扩增的 cDNA 快速高效地构建测序文库,DNA 片段化和接头连接在同一步完成,使用 Illumina 双接头策略。
- 扩增并测序。对带接头的 DNA 进行扩增并测序。
与 SMART-seq 的差异:
- SMART-seq 是在42度下进行RT,但由于部分 RNA 具有二级结构,出现位阻现象,导致链的延伸提前终止。SMART-seq2 中加入了甜菜碱,是一种甲基供体,能够增加蛋白质热稳定性。SMART-seq2 先在50度下反应2 min,破坏 RNA 二级结构,然后回到42度,进行2 min RT,循环多次,提高cDNA产量
- 甜菜碱的加入需要提升氯化镁浓度。镁离子能与甜菜碱中的羧酸阴离子结合,作为缓冲液帮助细胞应对渗透压的变化。有报道称过量氯化镁可能对 PCR 保真性有负作用,但在 SMART-seq2 的 RT-PCR 以及 PCR 阶段并未发现
- SMART-seq 使用的 TSO 3’端是3个 riboguanosines,并使用 Moloney murine leukemia (M-MLV) 逆转录酶,后者能够进行模版转换,合成 TSO 互补链;SMART-seq2 中前两个碱基还是 riboguanosines,第3个是修饰过的鸟嘌呤得到的 locked nucleic acids (LNA)。LNA 单体具有更强的热稳定性,以及在退火阶段高效结合到cDNA非模版3’端。RT 酶则换成了 Superscript II (Invitrogen),它是 M-MLV 编辑得到的,Rnase H 酶活性降低,热稳定性增强
- SMART-seq2 预扩增时用 KAPA HiFi HotStart ReadyMix 替换了 Advantage 2 polymerase mix,取消了先用磁珠富集 cDNA 这一步,简化了 protocol
CEL-seq (2012)
CEL-seq 通过体外扩增(In vitro transcription, IVT)实现单细胞测序。
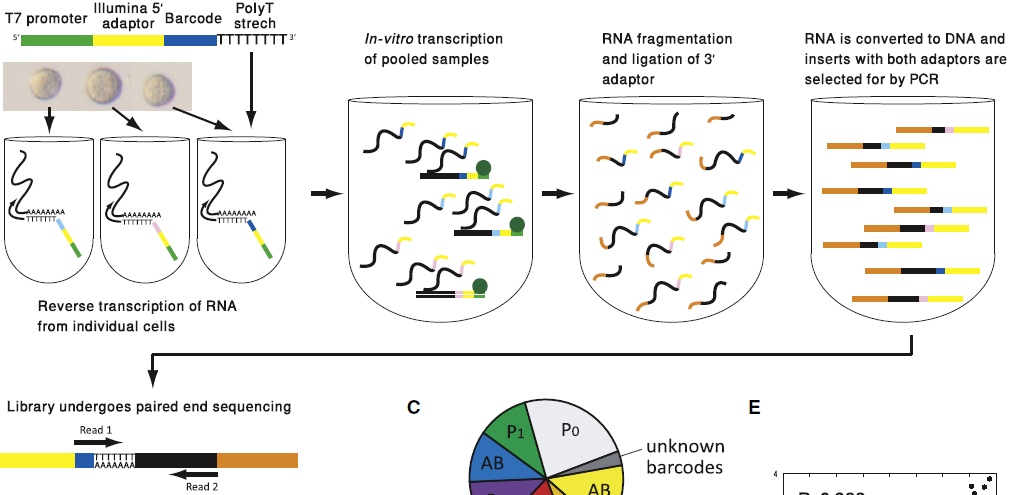
- RT。引物中包含 poly(T),barcode,5’ Illumina 测序接头和 T7 启动子,结合到具有 ploy(A) 尾的 mRNA 上,合成 cDNA。
- 合成第二条链后,形成的就是双链的 DNA。将所有的 cDNA 样本混合到一起,达到足以进行 IVT 的模版量。T7 promoter 用于启动IVT,以第二条合成的 DNA 为模版转录 RNA(所以此时的 RNA 序列与原 mRNA 序列是反向互补的,即 antisense RNA,aRNA),此时5’端最外为 Illumina 5’adaptor。经过多轮 IVT,实现模版的线性扩增
- 扩增后的 RNA 直接进入 RNA 模版准备步骤,被打断适合测序的大小,连接上 Illumina 3’ adaptor
- RNA 反转录成 DNA。两端都具有接头,且含有 barcode 的片段被用于双端测序。read1 记录 barcode 信息,read2 用于确定 RNA
CEL-seq2 (2016)
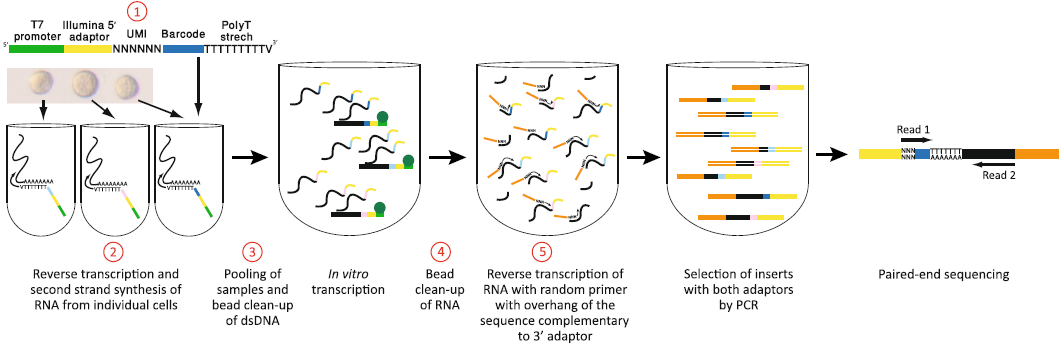
相较 CEL-seq,做出了一些改进:
- RT 引物中加入了一段6nt的 UMI,将8nt 的 barcode 缩短至6nt,并缩短 T7 引物和 Illumina 5’ 接头,使引物总长从92nt缩短至82nt,提升了 RT 效率
- RT 时使用 SuperScript II,并在第二条链合成时使用 SuperScript II Double-Stranded cDNA Synthesis Kit
- 改进了去除 dsDNA 和 aRNA 的过程,提高产量
- IVT 得到的 RNA 在反转录时,直接插入接头,不需要进行连接
Drop-seq(2015)
首先是构造大量不同的 barcoded beads,示意图如下:
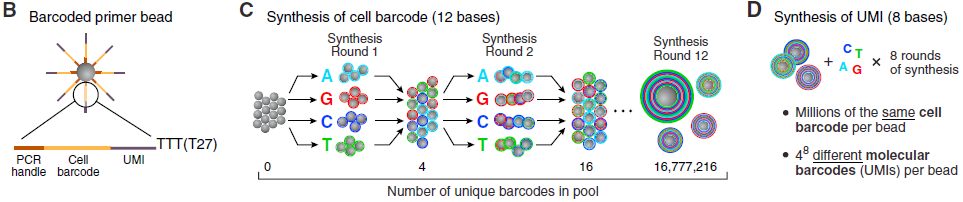
直接在每个 beads 上从5’-3’合成寡核苷酸引物,3’用于启动逆转录。每条寡核苷酸链包含4个部分:
- PCR handle,一段共有的序列,作为 PCR 和测序的起始位置;
- 细胞标签,用于标记细胞身份,同一 bead 中均相同;
- UMI,用于排除 PCR 偏好性带来的干扰
- oligo-dT 序列,捕获带有 poly(A) 尾的 mRNA,起始 RT
barcode 的合成采用 split-and-pool 策略,将上百万个磁珠分装成4等份,分别加入A,G,C,T,然后全部混合,再随机封装,共计循环12次,得到足够多的不同barcode;之后整个微珠池进行8轮的简并寡核苷酸(degenerate oligonucleotide)合成,得到 UMI
然后让我们来看一下后续的整个分析过程示意图:
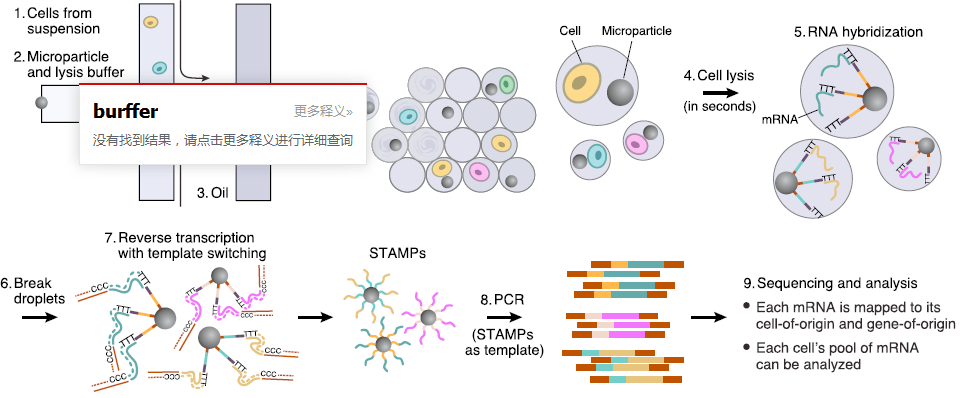
- 设备中两条通道分别导入含有微珠的细胞裂解液和细胞悬液,穿过油通道,形成微滴
- 在微滴中裂解细胞,mRNA 结合到磁珠上,形成 STAMPs (single-cell transcriptomes attached to microparticles)
- 加入试剂破坏水油表面,使得微滴破坏,所有的 STAMPs 混合到一起,进行 RT(使用的是模版转换来获取全长 cDNA),生成共价结合,稳定的 STAMPs(cDNA 已经与磁珠上的引物连接在一起了)
- 之后就对 cDNA 进行 PCR 扩增,然后进行测序
inDrop-seq(2015)
首先来看 barcoded hydrogel microspheres(BHM,带条形码的水凝胶微球) 的合成过程,示意图如下:
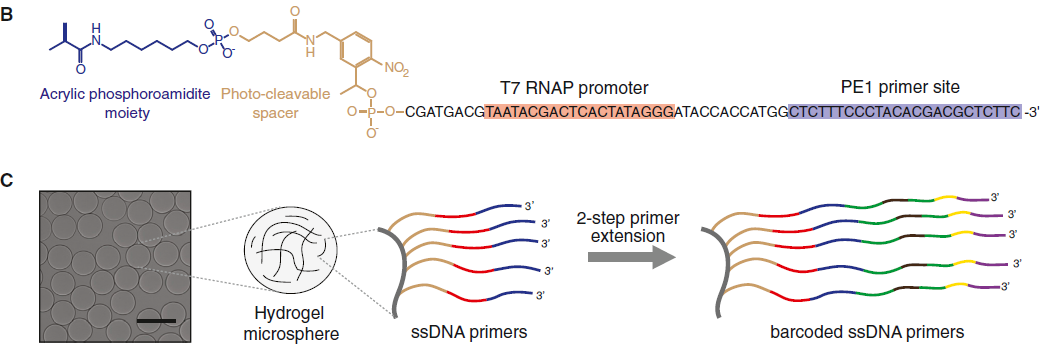
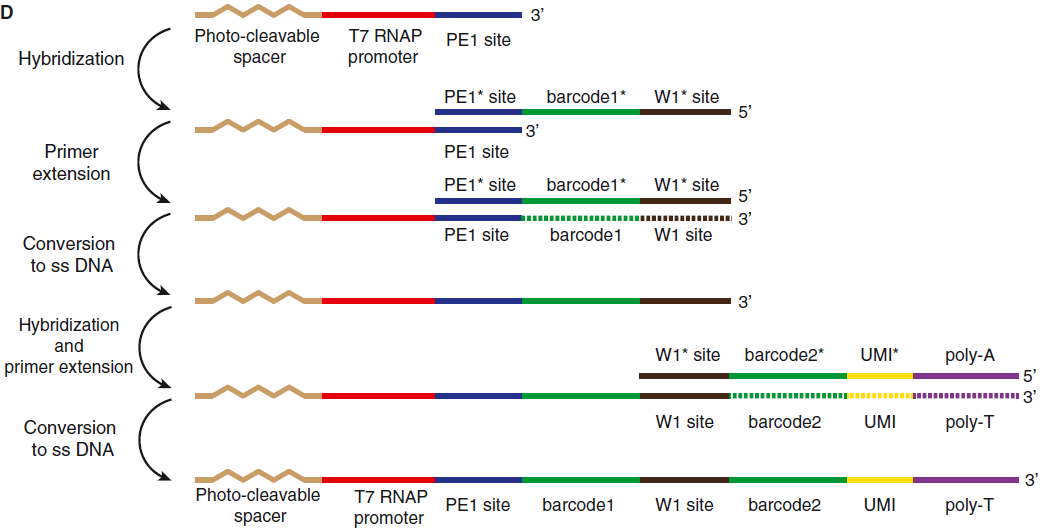
其中看上图B中的结构,acrylic phosphoroamidite moiety 是常用的 DNA 引物,后面是一段可光解的间区和 T7 RNA 聚合酶启动子序列,以及测序引物。之后,经过两部的引物扩展(图D,*代表反向互补序列):先进行引物延伸,加上了 barcode1 和 W1 site,然后转换成单链 DNA,再进行一次杂交和延伸,加上 barcode2,UMI 和 poly(T)
再来看整一个测序的过程:
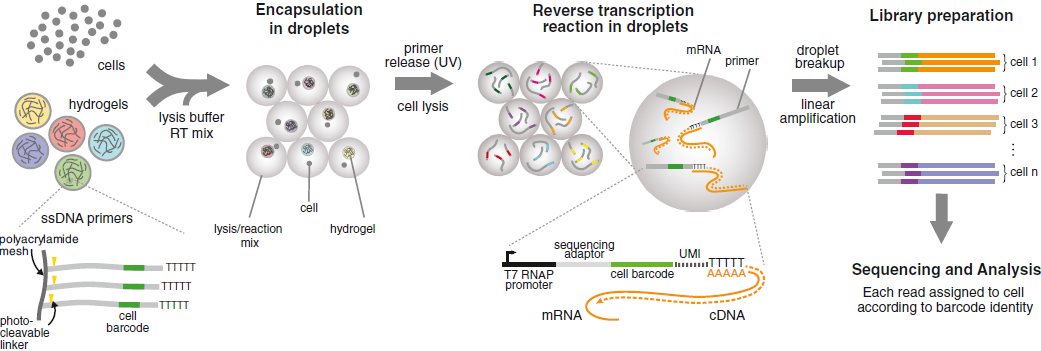
- 在微流设备中有4个输入通道,分别是 BHMs,细胞,RT/lysis 试剂以及油,封装成微滴
- 利用 UV 释放 BHMs 上的引物,与裂解细胞得到的 mRNA 结合,进行 RT
- 破坏微滴后将所有 cDNA 整合,按照 CEL-seq protocol 进行测序
CITE-seq (2012)
Cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq) 利用寡核苷酸标记的抗体,对细胞表面蛋白和和转录组同时进行测定,其流程如下图所示:
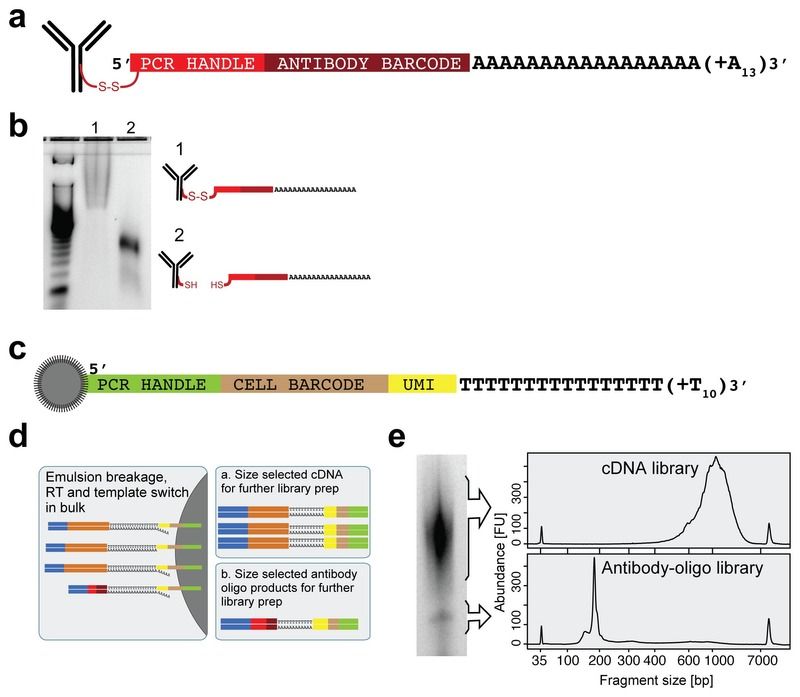
- 如图a所示构造 DNA-barcoded antibodies。通过链霉亲和素-生物素互作,将寡核苷酸5’端与抗体相连,而这种通过二硫键的结合会在还原条件下断裂,释放寡核苷酸(图b所示)。寡核苷酸中包括用于 PCR 扩增的 handle,用于抗体身份识别的 barcode,以及能与 oligo-dT 引物结合的一段 polyA 序列。
- 将 antibody-oligo 复合物与单细胞悬液共孵育,条件设置参照流式细胞染色,之后将未结合的抗体移除,然后进行 scRNA-seq。
- 以 Drop-seq 为例(beads 构成如图c所示),在微流体设备中将单细胞封装成小液滴,然后细胞裂解,使得带有 oligo-dT 的 beads 与细胞 mRNA 及抗体-寡核苷酸复合物结合,然后对同一细胞的 mRNA 和寡核苷酸进行 RT 和 PCR(图d),并标记上相同的 cell barcode。
- 扩增得到的 cDNA 和 antibody-derived tags (ADTs) 可以通过大小进行区分,从而分别进行 Illumina 测序文库的制备。由于两种文库可以独立生成,因此可以调整其在单个 lane 中的比例以确保各自所需的测序深度。
REAP-seq
RNA expression and protein sequencing assay (REAP-seq) 实现了在单个细胞中同时测定蛋白质和 mRNA。细胞通过连接了 DNA barcode 的抗体进行标记,其中抗体 DNA 标签的设计示意图如下所示:
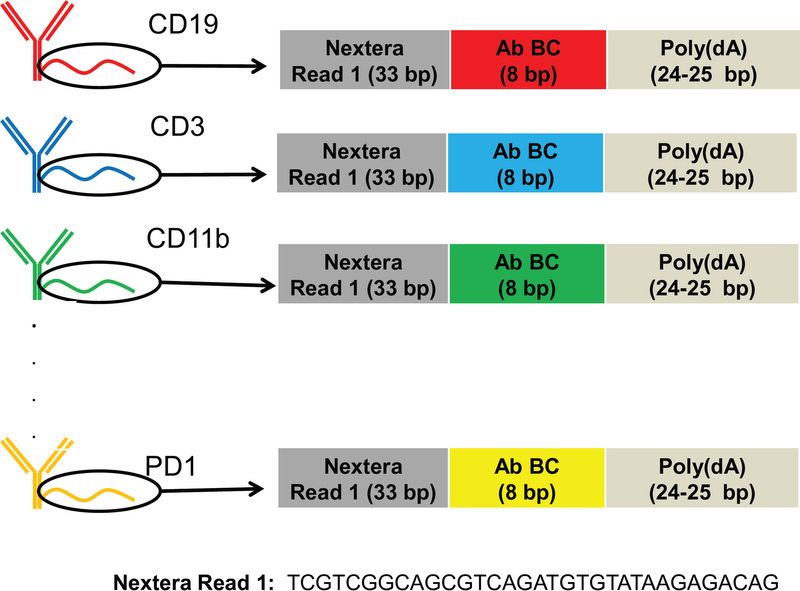
可以看到抗体与长为65-66bp的寡核苷酸偶联。寡核苷酸包含3个部分:(1)33 bp Nextera Read 1 sequence,作为后续扩增和测序的引物;(2)8bp长的独一无二的抗体标签(antibody barcode, Ab BC);(3)24-25 bp poly(dA) sequence。
REAP-seq 测序流程如下所示:
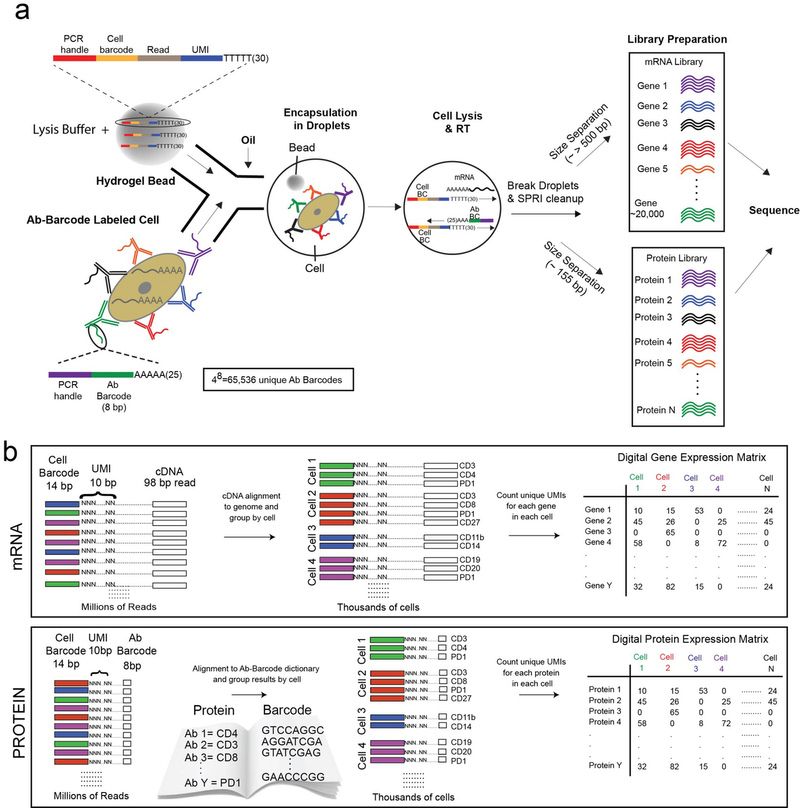
- 将 antibody DNA label 与细胞共孵育,之后洗脱未连接的 AbBs。
- 如图a所示,将标记了抗体-DNA 标签的细胞用 10x Genomics single-cell (sc)RNA-seq platform 进行处理。通过 DNA 聚合酶实现同时扩增 AbBs 和 mRNA,并利用核酸外切酶Ⅰ将过量的未结合的单链寡核苷酸消化,防止不同细胞间的 barcode 与 AbBs 结合。
- 破坏液滴后通过大小对 AbBs 和 mRNA 进行分选,然后制备文库和测序。
REAP-seq 与 CITE-seq 的差异在于前者在抗体和胺化了的 DNA barcode 之间构建了稳定的共价键,能够减小空间位阻。减小空间位阻对于蛋白试验的扩展性至关重要,将来也能拓展至细胞内标记。
各步骤实现的方法
总的而言,scRNA-seq 相较 bulk 要解决的主要是以下两个问题:(1)单细胞的分离;(2)RT 以及(3)cDNA 的预扩增
首先来看单细胞的分离:
- 酶处理
- 激光捕获显微切割(laser capture microdissection,LCM)从冷冻切片上切除细胞,低通量,劳动密集型,获得完全干净的细胞在技术上也有挑战性
- 膜片钳(patch clamping)
然后是单细胞的分选和捕获,主要有三大类:
- 微孔板(microwell):通过微移液枪或者激光捕获将细胞分离至微孔中
- 能与流式细胞仪结合,从而快速将单个细胞分选至微孔内,此外还能够通过荧光标记富集特定细胞群
- 能够检查每个孔内是否存在以破坏的细胞或者 doublets
- 相较于微流系统,microwell 对细胞的大小没有严格的限制
- 主要缺点是它不能像微流方法一样将反应缩减至纳升级体积,从而导致每个细胞平均的试剂消耗较高
- 微流体(microfludic)中的微室(microchamber):Fluifigm C1 微流系统,通过 IFC 芯片一次性捕获800个细胞,并在微室中完成细胞裂解,RT 和扩增
- 通量高于微孔板,且能获得高质量的表达谱
- 只有约10%的细胞会被捕获,因此不适合用于处理原材料有限的情况
- 有几种可选的细胞大小范围,要求细胞大小均一,对具有粘性或者非球形的细胞的捕获效率较低
- 每个 IFC 芯片的价格较贵,但由于能将反应体积缩小至纳升级,减少了试剂的消耗
- 微流体(microfludic)中的微滴(microdroplet):将细胞包裹在纳升级的微滴中,以微滴为反应器构建文库
- 能够捕获成千上万个细胞,将反应缩减至纳升甚至皮升级别,减少试剂消耗,适合大量细胞的分析
- 10X genomics 公司的 Chromium 系统相较 Drop-seq,在每个细胞内能够检测到更多的基因,能对更多的输入细胞进行测定,且设置和操作更简便,缺点是耗材价格更高。Drop-seq 中,细胞和 bead 是以随机的形式混合到微滴中的,因此需要进行足够程度的稀释才能避免混入两个细胞或两个 bead。对于 Drop-seq,其目标是在没20个微滴中捕获1个细胞,每10个液滴捕获1个 bead。所以,该系统经历双重泊松分布,只有约5%的输入细胞会产出转录组数据。相反,Chromium 系统产生的每个微滴几乎都只有单个 bead,故只需经历单个泊松分布,结果是约50%的输入细胞能产出转录组数据,远高于 Drop-seq
之后是反转录过程中,有两种主要的方法:
- poly(A) or ploy(C) tailing,在 cDNA 5’端加上通用接头以便后续的 PCR 扩增
- template switching,能够获得全长转录本,减少由于 RT 提前终止导致的 3’ coverage bias
cDNA 的预扩增也有两种方法:
- PCR,指数扩增,效率取决于序列,偏向 shorter and less G-C rich 的序列
- in vitro transcription(IVT),线性扩增,省去了模版转换步骤,但最后还需要额外的一轮 RT,可能导致额外的 3’ coverage bias
各 protocol 之间的系统比较
- Ziegenhain et al (2017)对比了 CEL-seq2, Drop-seq, MARS-seq, SCRB-seq, Smart-seq 和 Smart-seq2 6种方法,发现:
- scRNA-seq 达到每个细胞1000,000条 reads 就达到了合理的饱和测序深度,足够进行后续的比较分析
- 敏感性方面,Smart-seq2 是这些方法中最敏感的,找到了最多的基因,且覆盖度最均一;之后是 Smart-seq/C1,CEL-seq2/C1,SCRBseq;最后是 Drop-seq 和 MARS-seq
- 准确性方面,6种方法之间的差异不大,均足够给出较准确的表达量
- 精确度方面,Smart-seq2 最佳,但是其受扩增偏好性的影响也要高于其他使用 UMI 的方法
- 综合上述各角度,检测效力上,SCRB-seq 在一百万和五十万条 reads 时最有效,CEL-seq2/C1 在测序深度为25万条 reads 时最有效
总体而言,Drop-seq 最适合大量细胞的低深度测序,SCRB-seq 和 MARS-seq 适合细胞较少时的情况
Spike-ins
Spike-ins 是一组外源 RNA,加入到细胞裂解产物中,与内源 RNA 一同进行后续的测序分析。每份 spike-ins 的数量是一样的,然后根据最终检测的表达量来量化技术性方差
Spike-ins 是目前量化技术噪音最好的手段,但在实际应用中也存在一些局限性:
- Spike-in 长度相对人类 mRNA 平均值较短,考虑到大部分 scRNA-seq 技术存在3’ coverage bias,较短的外源 RNA 可能带来问题
- Spike-in RNA 分子的 poly(A) 相较内源 mRNA 较短,且没有5’帽子结构,RT 效率可能存在差异
- 内源 mRNA 可能存在二级结构或者结合蛋白,从而导致转换效率较低
- Spike-in RNA 分子的 GC 含量与内源 mRNA 也存在差异,可能带来干扰
总体而言,ERCC spike-ins 的捕获效率相较内源 mRNA 较低,另外某些情况下,ERCC spike-ins 的技术性方差要高于内源基因
UMI
UMI (unique molecule identifier) 是一段核苷酸序列,在 RT 过程中添加到 cDNA 上,每个分子携带独一无二的 UMI,用于消除 PCR 偏好性带来的干扰,主要能够提升低测序深度时的效力
挑战
- 每个细胞内原材料较少,因此现有的方法都需要进行 cDNA 的预扩增,但是扩增偏好性导致结果与实际细胞中的分布情况并非完全成比例。为了克服这一问题,目前大部分技术都会结合 UMI 技术
- 从器官或者组织中分离出单细胞的过程可能会改变细胞的表达谱,例如有研究发现早期应答基因(early response genes)会在分离几分钟内迅速上调,所以在数据分析的过程中需要注意是否会干扰结果的判定。克服这一问题有以下几种方法:
- 对细胞核进行 RNA 测序。该方法目前用于尸体解剖样本(胞质 RNA 丢失)和肌肉与神经元细胞(过大导致无法使用 Fluidigm C1 系统)的转录组分析。不过该方法的问题是只有约10-20%的细胞 RNA 存在于细胞核中,且大部分转录本未经过处理,还含有内含子。细胞核中的 RNA 能否代表细胞质中的 RNA 仍然存疑
- 使用转录抑制剂。如通过添加 α- amanitin 抑制 RNA 聚合酶Ⅱ减少细胞分离过程中引入的人为干扰,但是细胞吸收 α- amanitin 较慢,且转录抑制剂不能阻碍 RNA turnover (RNA 降解)
- 使用适应寒冷的蛋白水解酶进行分离,从而在低温下进行操作,尽可能保留转录表达谱
- 另外就是细胞分离难易程度不同,细胞脆弱程度不同,所以最终的单细胞悬液很难完美地代表实际的细胞分布情况
- 生物学噪音如基因表达的波动性,细胞周期状态等