在单个条件,技术或者物种中,scRNA-seq 计算方法能够成功地发现和鉴定细胞表型。但是,跨数据集的细胞亚群的鉴定仍极具挑战性。在本文中,作者基于不同 scRNA-seq 数据集中共有的变异来源,实现对不同数据集中共有的细胞群体的鉴定以及下游的比较分析,并在 Seurat 包中实现了该方法,从而实现不同 scRNA-seq 数据集的一般化比较,有望加深我们对不同细胞状态对干扰,疾病以及进化等应答的理解。
Introduction
scRNA-seq 在诸多领域大放异彩,加深了我们对生命的理解。现有许多方法都能够有效地分析单一条件,技术或者物种的 scRNA-seq 数据集,但是针对不同的 scRNA-seq 数据集,例如在不同条件下比较异质性组织,或者整合不同测序技术得到的结果,仍有很大的挑战性。例如对 scRNA-seq 数据进行比较分析时,由于不同样本中各细胞类型所占比例发生了变化,以及对给定细胞类型而言,表达情况也有所不同,因此将多个数据集放在一起分析时,会导致两种本不相干的效应相互干扰。因此我们需要新的方法,能够整合不同的数据集,尤其是考虑到未来会有越来越多的数据。
本文献中,作者受计算机视觉中为校准和整合图像数据所使用的技术启发,提出了一种新的用于 scRNA-seq 数据整合分析的策略。这种为“流形对齐”所设计的多元方法被成功应用于 scRNA-seq 数据,找出不同数据集间保守的基因-基因相关模式,从而将细胞映射至一个共享的低维空间,实现整合分析。
Seurat 校准流程概观
为了能够比较不同条件,技术或者物种得到的 scRNA-seq 数据集,新的计算方法必须能够做到以下几点。为了更好的说明,作者以对异质性细胞群体用药和不用药时得到的 scRNA-seq 数据集为例(下图):
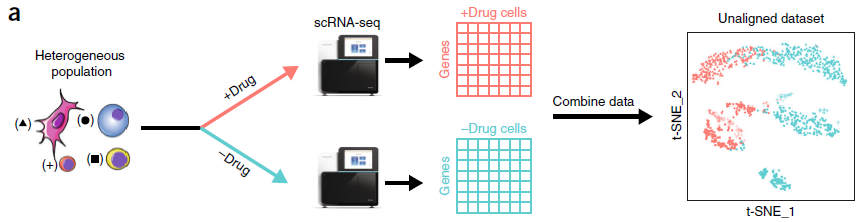
- 同一亚群的细胞应当对齐,即便它们有不同的药物应答反应;由于传统针对混池数据的批次效应校准方法假设某一因素对单个细胞群中所有细胞的影响是统一的,因此不适用于该情况
- 允许不同条件下细胞密度,即各亚群所占比例发生变化
- 能适应不同条件下特征的尺度范围的变化,不管是整体性的转录本变化,还是不同测序技术得到的数据集在标准化策略上的差异
- 该过程不应针对已知的细胞类型,不需要预先构建用于匹配各亚群的标记集合
Seurat 对齐流程主要包括以下几步:
- 利用
canonical correlation analysis (典型关联分析, CCA)不同数据集间共有的基因关联结构 - (可选)找出无法用这个共享结构进行很好的描述的单个细胞。这可以找出那些不同数据集之间未重叠的罕见细胞,做个标记用于后续进一步的分析
- 将数据集映射到一个保守型低维空间,使用非线性规整算法对特征尺度差异进行标准化
- 对整合的数据做下游分析,例如通过聚类区分不同的细胞亚群,或者重构连续性发育过程
- 对不同数据集对齐后的亚群做比较分析,从而确定群体密度或者基因表达的差异
找出不同数据集之间共享的关联结构
用于数据融合的机器学习算法旨在将来自多个实验的数据整合,以一个统一的形式表示,例如 CCA 的目标是找到数据集间相关性最强的特征的线性组合,将其定义为数据集间共享的相关结构 (shared correlation structure)。本研究中,作者基于同一组基因,利用 CCA 来确定不同数据集细胞之间的关系。除了将 CCA 用于两个数据集的整合外,还发展出了 multi-CCA 用于多个数据集的整合。此外,作者还采用了 CCA 的一种变形,对角线 CCA (diagonal CCA),用于细胞数多于基因数的情境。
理论上,计算过程中可以考虑任意一个所有数据集均测到的基因。不过实际情况中,集中于那些至少在某个数据集中呈现出高变异的基因。CCA 找出一组基准向量,然后将各个数据集的细胞映射到这个低维空间,从而使得不同数据集间,沿着这些基准向量(基因水平的映射)的变异高度相关。
对齐来自 CCA 的基准向量
CCA 返回的向量在基因水平的映射在数据集之间相关,但不一定对齐。除了需要线性转换用于校正特征尺度或归一化策略的整体性变化,我们同样需要非线性转换来校正群体密度的变化。因此作者对不同数据集的 CCA 基准向量进行对齐,得到单个整合的低维空间。简单讲就是将每个基准向量中表达与基准向量最相关的一些基因取表达量的加权平均,作为一个 metagene。之后,对这些元基因做线性转换,校正特征尺度的整体性差异;接着通过动态尺度规整确定元基因之间的映射,即在对其过程中局部压缩或拉伸向量,从而校正群体密度的变化。通过这些步骤,最终得到单个对其的低维空间代表所有的数据集,用于后续的整合性分析,包括无偏差聚类和重构发育轨迹。
激活的和静止的 PBMCs 的比较分析
作者首先通过包含多种受激和未受激的不同类型细胞的数据集来展示对齐策略的效果。最近有一项研究将来自8位病人的14039个 PBMCs 分成两组,一组用 IFN-β 刺激,另一组作对照,然后进行 scRNA-seq。由于所有细胞均有应答 IFN-β 的机制,所以产生的应答激烈但又呈现出高度的细胞特异性,从而导致聚类结果令人困惑,因为细胞同时按照类型和刺激条件聚集成簇(下左图)。最后,那篇文献的作者利用已知的标志基因将细胞分成8类。
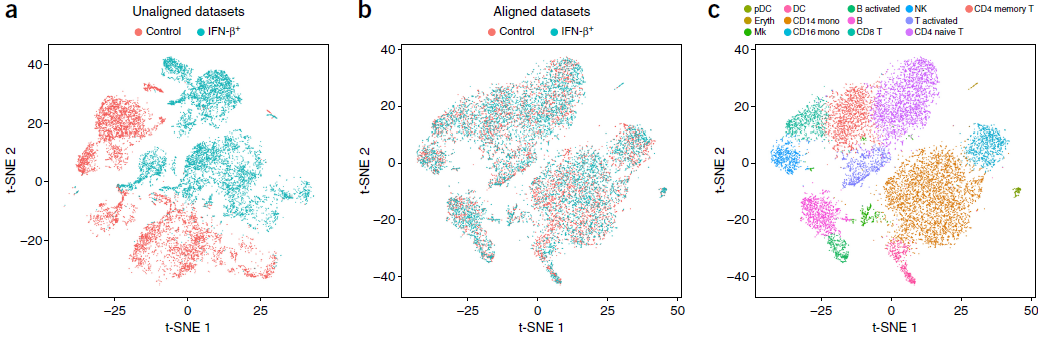
本文作者利用 Seurat 包获得一组典型相关向量,从而排除刺激条件的干扰将 PBMC 分成多个亚组(上图中),最终一共鉴定出13个细胞簇(上图右),囊括了之前研究得到的所有8类细胞。通过检测经典的细胞类型标志基因,对这些细胞簇的身份进行了验证(下图左部分)。
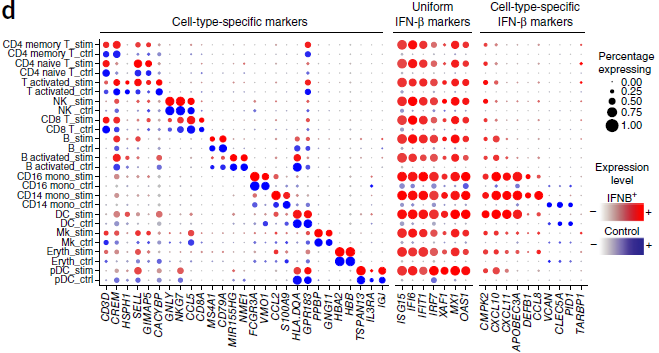
对齐数据集后,作者研究了不同 PBMCs 对 IFN-β 刺激的应答反应。由于不同条件组的细胞来自于同一个细胞池,因此两组中各细胞簇所占比例基本相同。每种类型的细胞受 IFN-β 刺激后均表现出明显的基因表达变化,我们可以看到(上图右部分)既有所有类型细胞均表达的一致性标志基因如 ISG15,IFIT1 等,也有不同细胞间存在差异的基因如 CXCL10 主要在骨髓细胞中被激活。
聚焦于最新发现的细胞簇,对比初始和记忆 CD4+ T 细胞对 IFN-β 的应答,发现应答特征几乎一致;但如果对比 conventional (DC) 和 plasmacytoid dendritic cells (pDC,浆细胞样树突状细胞) 的应答,发现两者之间明显的差异(下图),这种差异在每位病人中均存在。
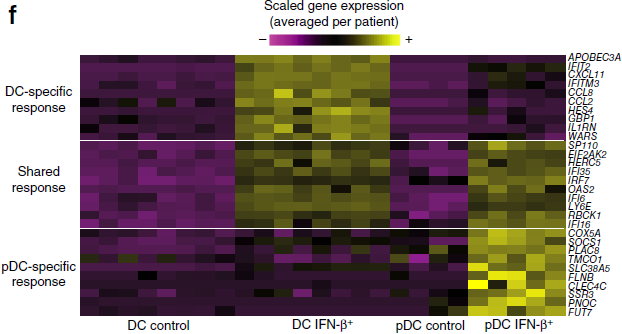
此外,比较所有细胞对 IFN-β 刺激的应答,我们发现骨髓细胞和淋巴细胞明显地聚集在一起(下图),而 pDC 则呈现出不同的应答反应,单独成簇。
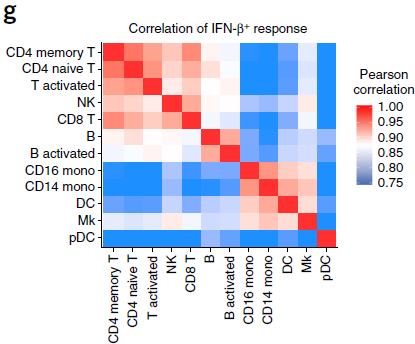
为了验证上述结论,作者重复了原始论文中的实验,利用标准表面标记将 DC 和 pDC 进行分类,然后对刺激组和实验组进行混池转录组测序,发现受 IFN-β 刺激而差异调控的基因在单细胞和混池数据集中呈现高度相似的特征。综上,在单个转录组分析中,该对齐方法能够通过整合性聚类准确地确定相同状态的细胞,并找出细胞特异性响应模块,这些模块可能在生物体对感染的免疫应答过程中起重要作用。
区分未重叠的群体
在之前的例子中,两个群体的细胞组成是一致的;因此,作者希望测试一下算法在数据集内存在非重复群体时的表现,因为那些群体特有的罕见细胞亚群,可能有着重要的生物学作用。为此,作者进行了两个计算机模拟实验,分别将前述受激组中的常见细胞(CD14+ 和 CD16+ 单核细胞,38%)和罕见细胞(有核红细胞,0.5%)移除,然后重复对齐步骤。当移除常见细胞群时,发现对整体聚类基本无影响,对照组的 CD14+ 和 CD16+ 单核细胞仍能够很容易地从聚类图中区分出来,只不过只在单个数据集中存在;但当移除有核红细胞时,其他亚群不受影响,但对照组的有核红细胞就难以独自成簇。因此,作者希望能够设计一个新的方法来确定这些未重叠的细胞,做上标签用于后续的探索。
作者推测 CCA 可能难以确定典型相关向量,用于定义仅在单个数据集中出现的罕见细胞,相反,PCA 可能可以区分出这些细胞。因此,他们对 CCA 定义的低维空间能够解释每个细胞的表达谱的程度做了量化,并与独立作用于各个数据集的 PCA 进行了比较。若 CCA 能解释的变异比例低于 PCA,表明存在数据集间未共享的变异。利用该方法,成功地分离出了有核红细胞亚群;而且利用该方法对原始的完整数据集和模拟的移除了单核细胞的数据集进行分析时,正确地没有对相似的细胞进行标记。
综上,整合分析对常见的未重叠群体足够强健,对单个数据集中出现的罕见细胞群,也能够区分出来用于后续分析。
不同 scRNA-seq 技术的整合分析
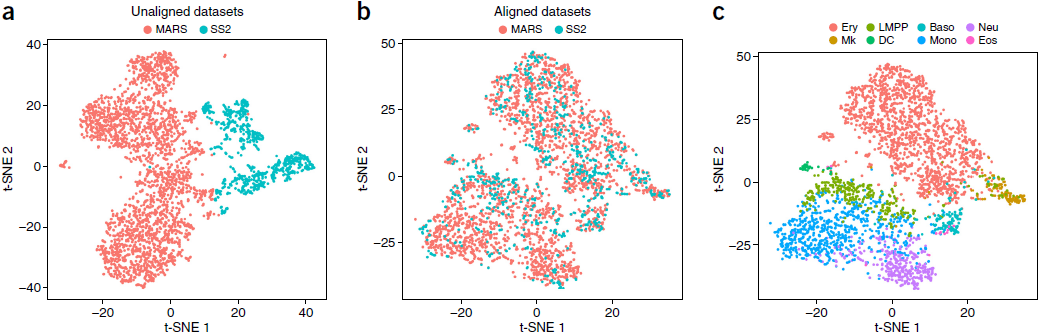
接下来,作者对同一组织通过不同 scRNA-seq 技术得到的数据集进行了分析。有两个研究组分别利用 SMART-Seq2 和 3’ MARS-Seq 对小鼠骨髓中的造血祖细胞进行了分析。不同的测序技术,标准化方法以及测序深度使得整合这两个数据集充满了挑战性。此外,两篇文献的着重点不同:SMART-Seq 数据的分析聚焦于绘制淋巴,骨髓以及红细胞的连续性轨迹;而 MARS-Seq 数据的分析则鉴定出18个不同的分支,隶属于8种不同的造血细胞系祖细胞。
Seurat 成功地从整合后的数据集中区分出不同的祖源细胞亚群(下图),与之前研究的结果一致。此外,还成功地找到了污染的 NK 细胞,标记为非重叠。
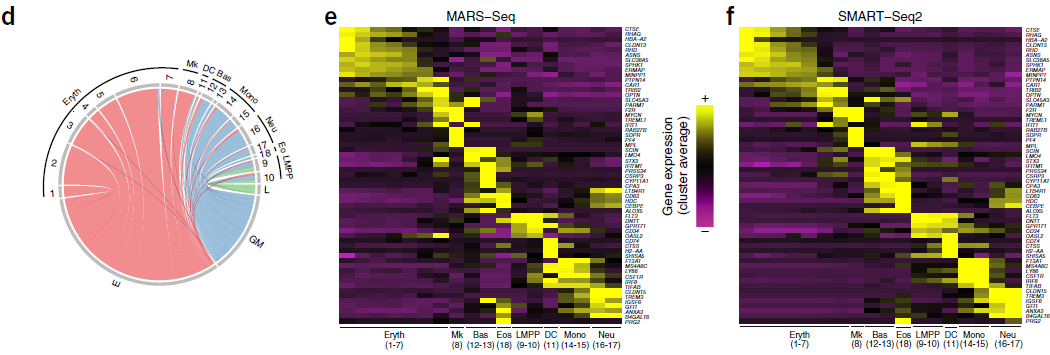
通过对齐,来自 SMART-Seq2 数据集的细胞被映射到 MARS-Seq 数据集中最相似的分支,例如 SMART-Seq2 中的早期巨核细胞-红细胞祖细胞与 MARS-Seq 中的红细胞和巨核细胞祖细胞(C1-7簇)对应;早期淋巴祖细胞对应淋巴成分祖细胞(淋巴源多能祖细胞,C9-10簇);粒细胞-巨核细胞祖细胞对应嗜碱性粒细胞,嗜酸性粒细胞,树突状细胞,嗜中性粒细胞和单核细胞祖细胞(C11-18簇)(下图左)。事实上,根据标记基因表达情况绘制热图,我们发现对齐后的两个数据的结果几乎一致(下图右),说明此次对齐的生物学驱动因素是谱系决定因子。
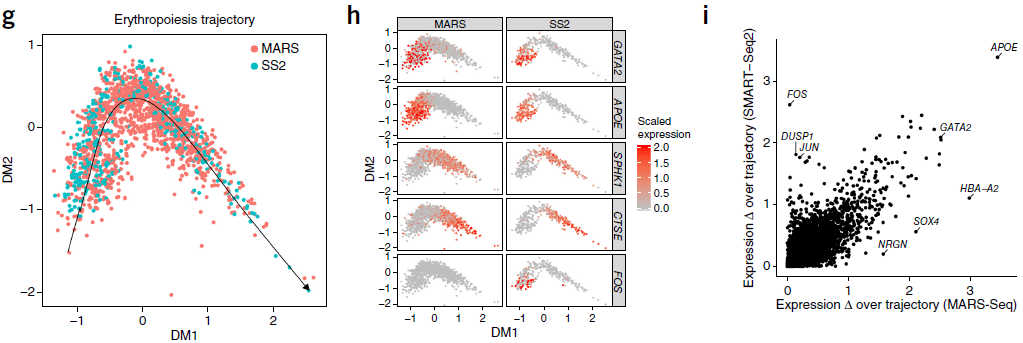
两个数据集都发现了红细胞分化过程中出现的发育异质性的亚群(MARS-Seq 中分为7个阶段,C1-7簇),因此作者将两个数据集中隶属于红细胞的细胞整合,重构发育轨迹(下图左)。这条发育轨迹既保留了两个数据集中细胞的拟时间分布顺序,也将两者对齐,使得两个数据集的细胞在典型的几个分化标志性基因的表达上呈现出几乎一致的变化过程(下图中)。此外,作者还发现基因的表达随着轨迹线的变化在两个数据集之间也是高度保守的(下图右,基本沿对角线分布),尤其是我们所熟知的那些红细胞生成效应因子。当然,也存在一些技术特异性效应,例如 scRNA-seq 过程中与细胞应激相关的 JUN/FOS 应答。综上,该算法仅能够实现离散和过渡态群体的对齐,也能够鉴定各个数据集之间保守或者特有的基因表达程式。
能够将同一异质性组织的数据进行整合使得我们能够联合来自不同实验室,采用不同技术得到的数据集进行 meta 分析。为了进一步验证,作者又加上了两个实例。一是整合来自4种 scRNA-seq 的人胰岛数据,成功鉴定出8种不同细胞类型。其中一种罕见的内皮细胞群虽然在所有数据集中均存在,但其过低的占比(1%)使得在最初大部分(3/4)的分析中未被发现。另外,考虑到每个样本来自于不同个体,样本中各细胞类型所占比例是高度差异化的,但也并未对整合产生干扰;第二个例子中,作者将三种不同技术得到的16653个人类 PBMCs 的数据进行整合,鉴定出16个免疫细胞群体,包括6种 T 细胞簇和一个罕见的 NK 细胞亚群(0.5%)。该亚群的罕见性使得它们未能在各个单独的数据集中被发现,包括一项囊括了68000细胞的分析。利用这个整合数据集进行基因差异表达分析,得到多于最初研究结果3倍的差异表达基因数量。
综上,整合不同测序技术得到的数据集能够增强统计效力,发现罕见细胞表型,并坚定各细胞状态的转录标志基因。
跨物种
最后一个例子中,作者对不同物种相同组织的数据进行了整合。数据来源于一项利用 inDrop 对人类和小鼠胰岛测序分别鉴定所包含的胰岛细胞类型的研究。该研究发现人和小鼠之间各类型细胞特异性转录组的保守性很差,难以找到物种间均存在的高表达标志基因。这些数据集的结构主要取决于物种类型和个体贡献者,广泛存在的差异给整合带来了明显的障碍。但是,作者仍认为存在一组基因-基因关联,从而将人类和小鼠细胞对齐。
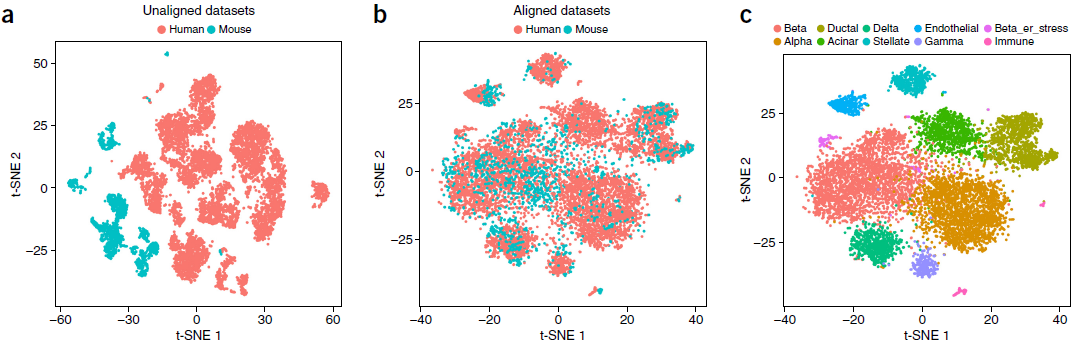
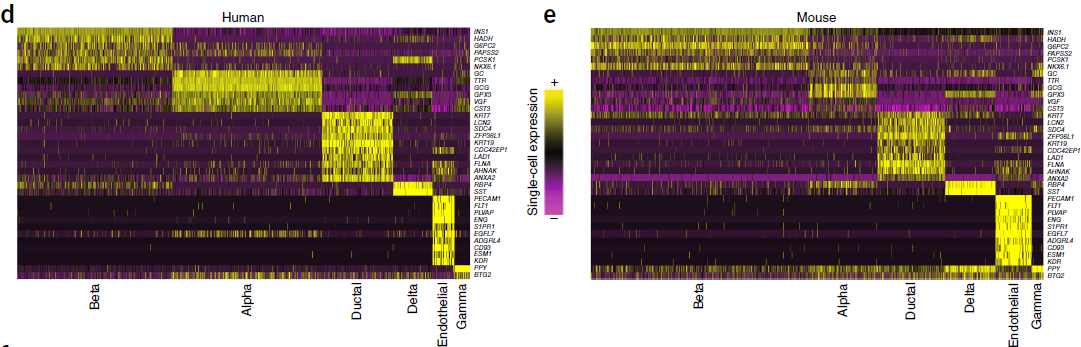
实际情况中,Seurat 确实能够将两个数据集很好地整合(上图),并将小免疫细胞群(人类肥大细胞和小鼠 B 细胞)标记为非重叠。进行聚类分析后,得到10个细胞簇。除了小部分(5.8%),特别是 UMI 数量低的那些细胞存在不一致外,其他与之前的研究结果非常吻合。此外,作者还找到了一组人和小鼠之间保守的细胞类型标志基因(下图)。
此外,作者还从人和小鼠中都找到了一种罕见的 beta 细胞亚群(下图)。与其他 beta 细胞相比,这些细胞 INS 表达量水平相同,但是内质网应激基因(HERPUD1;GADD45A)的表达上调。
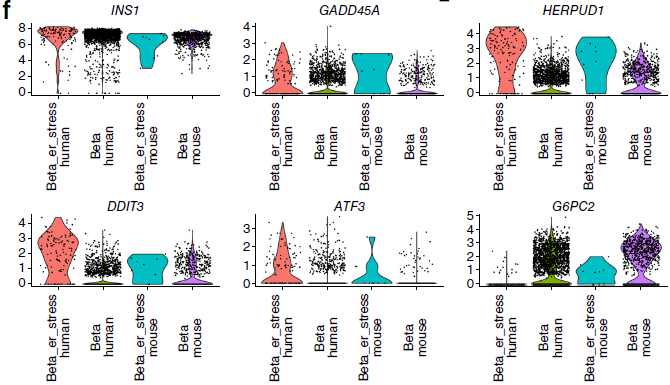
原研究在对人类 beta 细胞进行半监督式分析时也找到过类似的信号,但如果使用无监督聚类或者在小鼠中均未发现;而本文中提出的整合分析则找到了一组保守性标志基因,富集于内质网对未折叠蛋白应激调控基因(下图)。这些调控基因在糖尿病发生发展过程中起重要作用。另外需要注意的是,转录因子 ATF3 和 ATF4 在两个物种中均高表达,而这些因子都是人们所熟知的在胰腺的应激反应起始中起重要作用。
综上,该整合方法能够从上万年进化导致的显著全局转录组变化中鉴定出共有的细胞状态。
对比其他对齐和批次效应校正技术
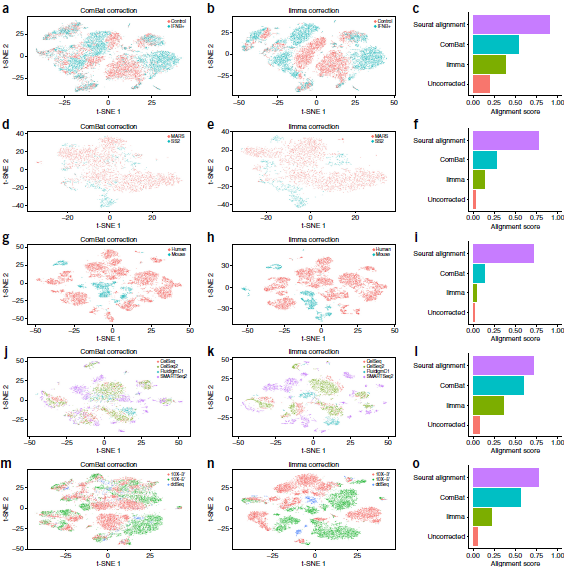
作者还将 Seurat 与其他在混池和单细胞基因组中广泛应用的批次效应校正软件进行了比较。为了量化分析每个软件,他们设计了一种被称为 alignment score 的指标,范围为0-1,来检查对齐后每个细胞的邻域:如果数据集被正确地对齐,那么邻域中应当包含相同数量的两个数据集的细胞。比较结果如下图,可以看到 Seurat 表现最佳,尤其是在数据集之间的转录组差异(如批次效应)明显胜过细胞类别(生物学)差异如跨物种整合时,表现差距更大。
Discussion
本文献中,作者通过识别共有的变异来源,对应不同实验中的亚群,对 scRNA-seq 数据集进行整合,并在 Seurat 包中实现了这一算法。数据整合是实现在单细胞层面对病例-对照研究普遍性框架的关键步骤。之前的研究发现,没有哪种 scRNA-seq 技术全面优于其他方法,每种方法都有其优势和缺陷,因此整合不同数据具有显著的潜在价值。
本文中,对于跨物种数据集的整合,算法也给出了正确的回应。这些分析可能为我们利用小鼠模型研究人类疾病提供了有用的比较工具,可能使得我们能够通过从小鼠中发现的致病群体找出人类中对应的群体,或者相反。此外,新的数据集使得能够跨物种对齐和比较发育轨迹,从而加深我们对导致细胞多样性的基因调控网络在进化过程中是如何布局的理解。
当然,仍有诸多问题有待解决。虽然本方法能够联合分析具有重叠和未重叠群体的多个数据集,但考虑到未来的数据集科能由成百上千个不同批次的组成,它们具有显著变化的大小和未重叠的群体,我们可能需要新的方法。此外,本文献中所使用的数据集较小,占用的资源有限,但未来包含上万细胞的数据集可能需要更先进的计算,抽样或优化的算法。
REF
- Butler A, et al. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nature Biotechnology. 2018, 36:411-420. doi: 10.1038/nbt.4096.